ABSTRACT
The goal of this work was to achieve fast determination of glucosamine in pharmaceutical formulations by high performance liquid chromatography. Commercial tablets were dissolved in water and after filtration, 6 μL were injected to Luna C18 column (250 mm × 4.6 mm, 5μm). Diode Array Detector (DAD) was set at 193 nm. Using sodium perchlorate (50 mM, pH 6.5): acetonitrile (99:1, v/v) at 0.8 mL min-1 as a mobile phase, glucosamine eluted with the retention time 2.834 min ± 0.003 min and the peak purity in real sample was 998.9 ± 3.3. The detection limits evaluated for peak height and peak area measurements were 0.05 μg mL-1 and 0.02 µg mL-1, respectively. The between-day precision relative standard deviation (RSD) evaluated at two concentration levels was always below 1%.
RESUMEN
El objetivo de este trabajo fue lograr la determinación rápida de glucosamina en formulaciones farmacéuticas mediante cromatografía líquida de alta resolución. Las tabletas comerciales se disolvieron en agua y después de filtración la solución (6 μL) fue introducida en la columna Luna C18 (250 mm × 4.6 mm, 5μm). La detección UV se llevó a cabo en 193 nm. La fase móvil fue perclorato de sodio (50 mM, pH 6.5): acetonitrilo (99:1, v/v) con el flujo total de 0.8 mL min-1. En estas condiciones, el tiempo de retención fue 2.834 min ± 0.003 min con la pureza de pico en la muestra real de 998.9 ± 3.3. Los límites de detección evaluados para altura y área del pico fueron 0.05 μg mL-1 y 0.02 µg mL-1, respectivamente. La precisión evaluada para dos niveles de concentración con una desviación estándar relativa (RSD, por sus siglas en inglés) fue siempre inferior al 1%.
INTRODUCTION
An amino monosaccharide glucosamine (2-amino-2-deoxyglucose) is widely used in the treatment of degenerative joint diseases (Chiusaroli et al., 2011; Distler & Anguelouch, 2006; Ragle & Sawitzke, 2012). It has also been accepted as a dietary supplement by the US Food and Drug Administration; however its effectivity is now under discussion (Vlad, LaValley, McAlindon & Felson, 2007). The natural source of glucosamine is chitin, typically obtained form sea shrimp and crab shells (López-Cervantes, Sánchez-Machado & Delgado-Rosas, 2007; Zhu, Cai, Yang & Su, 2005). The producion of pharmaceuticals and dietary supplements involves the liberation of glucosamine from chitin by means of acid hydrolysis; poor control of glucosamine content in commercial products has been highlighted as an important cause of inconsistent outcomes in clinical trials (Aghazadeh-Habashi & Jamali, 2011). In this regard, analytical control of glucosamine in the commercial products becomes an issue. Several procedures are available for this purpose, including simple spectrophotometric assays (Gaonkar, Khanvilkar, Shettigar & Gadgoli, 2006), thin layer chromatography (Solomon, Kumar, Anand, Sivakumar & Venkatnarayanan, 2010), liquid chromatography (LC) or capillary electrophoresis (CE) separations with spectrophotometric, contactless conductivity, amperometric or fluorimetric detection (Ander, Karlsson & Ohrlund, 2001; Campo, Campo, Ferlazzo, Vinci & Calatroni, 2001; Chaisuwan et al., 2011; Cheng & Kaplan, 2003; Qi, Zhang, Zuo & Chen, 2006; Soga & Heiger, 1998). Among various studies reported previously, the most common approach relies of suitable derivatization followed by LC separation with spectrophotometric or spectrofluorimetric detection (Anumula, 1994; Cohen & De Antonis, 1994; Domínguez & Dunn, 1987; Eikenes, Fongen, Roed & Stenstrømc, 2005; Liang, Leslie, Adebowale, Ashraf & Eddington, 1999; Palace & Phoebe, 1997; Yu, Cai, Zuo & Duan, 2005; Zhou, Waszkuc & Mohammed, 2004; 2005; Zhu et al., 2005). The derivatization agents used so far have been: o-phtaldialdehyde (OPA) (Eikenes et al., 2005; Nemati, Valizadeh, Ansarin & Ghaderi, 2007) 9-fluorenylmethoxycarbonyl chloride (Fmoc-Cl) (Zhu et al., 2005), 9-fluorenylmethyl-chloroformate (López-Cervantes et al., 2007), N-(9-fluorenyl-methoxycarbonyloxy) succinimide (FMOC-Su) (Zhou, Waszkuc & Mohammed, 2004; 2005), dansyl chloride (Qi et al., 2006) 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (Cohen & De Antonis, 1994; Palace & Phoebe, 1997), 2-aminobenzoic acid (Anumula, 1994) and phenylisothiocyanate (Liang et al., 1999). Even though pre-column derivatization enables for enhanced selectivity and sensitivity of determination, it requiers additional procedural steps and makes the whole procedure less attractive for routine analytical applications. To meet the desired simplicity and speed, the direct chromatographic method has been proposed by Shao, Alluri, Mummert, Koetter & Lech (2004). In that work however, the separation of glucosamine was achieved on the specific amine column and the separation time was 15 min.
In the present study, the direct determination of glucosamine has been undertaken, aiming the development of a new, simple and fast reversed phase chromatographic procedure, suitable for routine analytical control of glucosamine-based pharmaceutical formulations.
MATERIAL AND METHODS
Instrumentation
An Agilent Series 1200 liquid chromatograph equipped with a quartenary pump, well plate autosampler, a diode array detector and Chemstation (Agilent Technologies, Palo Alto, CA, USA). The chromatographic column was Luna C18 (250 mm × 4.6 mm, 5μm) from Phenomenex.
Reagents and samples
All chemicals were of analytical reagent grade. Deionized water (18.2 MΩ cm, Labconco, USA), HPLC grade acetonitrile and methanol (Fisher Scientific, Pittsburgh, USA) were used throughout. D-(+)-Glucosamine hydrochloride (99.9% purity) from Sigma was used as a standard. The following Sigma reagents were used: hydrochloric acid, perchloric acid, boric acid, sodium hydroxide, sodium acetate and o-phthalaldehyde (OPA). Three commercial pharmaceutical formulations were purchased in the local market.
Procedures
Standard/sample preparation
A stock solution of glucosamine was prepared by weighing precisely 900 mg of the Sigma reagent and dissolving in 100 mL of deionized water (ultrasonication for 15 min). Working solution corresponding to the highest calibration standard (0.9 mg mL-1) was prepared daily by appropriate dilution. For each pharmaceutical product, 25 tablets were weighed and ground in porcelane mortar. Five aliquots corresponding to the average mass of one tablet were precisely weighed and transferred into a volumetric flasks. For better solubilization, each suspension was ultrasonicated (10 min) and the volume was brought to 100 mL with deionized water. The samples were centrifuged (10000 g, 10 min), diluted 1:10 (v/v) with the mobile phase and introduced into the HPLC autosampler vials.
Reversed phase separation without pre-column derivatization
Isocratic elution was carried out with the mobile phase composed of sodium perchlorate (50 mM perchloric acid adjusted to pH 6.5 with 1 M sodium hydroxide) – acetonitrile (99:1, v/v) at a flow rate 0.8 mL min-1. The UV detection was at 193 nm (bandwidth 6 nm) and the injection volume 6 μL. The column was termostated (20 °C), the chromatographic run was set at 4 min and the next injection was performed with no need for column cleanning and equilibration. The calibration was performed in automatic mode, setting the concentrations at 0 mg mL-1; 0.15 mg mL-1; 0.30 mg mL-1; 0.60 mg mL-1; 0.90 mg mL-1. For recovery experiments, the samples were spiked also in automatic mode. In brief, the sample volume was set at 6 μL, then 0 μL, 4 μL and 8 μL of standard solution (0.5 mg mL-1) were added, the volume was brought to 20 μL with the mobile phase and injected.
Reversed phase separation after OPA derivatization
The derivatization of glucosamine in the calibration solutions and in the real samples was performed in the automatic mode.The following steps were programmed: (1) draw 5 µL of borate buffer (0.4 M, pH 10); (2) draw 5 µL of methanolic OPA solution (40 mM); (3) wash needle, 1 s; (4) draw 6 µL of sample (or calibration solution); (5) draw mobile phase to complete 20 µL; (6) mix in washport, 6 times at maximum speed; (7) needle wash in the flush port, 1 s; (8) wait 2 min and (9) inject. The mobile phases A – acetonitrile, B - methanol, C – sodium acetate (30 mM, pH 7.0) were used in the following gradient: 0 min 15% A, 5% B, 40% C; 7.0 min 50% A, 5% B, 15% C; 7.5 min 70% A, 5% B, 15% C; 8.5 min 40% A, 5% B, 20% C; 9.5 min 15% A, 5% B, 40% C. Total flow rate was 0.8 mL min-1, column was thermostated at 30 °C and the UV detection was at 340 nm. The calibration range and the sample preparation was the same as in the procedure without derivatization.
Statistical analysis
Descriptive statistics was performed to obtain means and standard deviations. To detect possible differences between the results obtained by two chromatographic procededures, ANOVA (Analysis of variance) was carried out using Statistica for Windows software (StatSoft Inc, Tulsa, OK). Significance level was established at p < 0.05.
RESULTS AND DISCUSSION
Chromatographic determination of non-derivatized glucosamine by reversed phase chromatography with spectrophotometric detection is hampered by its high polarity and the lack of absorption band in visible wavelength range; however this particular approach would be quite attractive for routine analytical applications. In this work, several buffering and/or ion-pairing reagents have been examined as components of the mobile phase in order to achieve acceptable retention of glucosamine on typical analytical column (Luna C18). Specifically, phosphate, borate and acetate sodium or ammonium salts, heptafluorobutyric acid and sodium perchlorate in the range of pH from 3.0 to 7.5 were examined. The effect of the mobile phase ionic strength has also been explored, using the concentration range of the above salts from 10 mM up to 50 mM. The proportion between aqueous solution and organic modifier (acetonitrile) has been tested from 99.5:0.5 to 95:5 (v/v). Finally, the flow rate has been varied in the range 0.5 mL min-1-1.5 mL min-1. In each case, the selection criterions were: short retention time of glucosamine with acceptable capacity factor and high peak purity. The conditions finally selected are presented in Materials and methods.
Considering the absorption spectrum of glucosamine in UV region, the detection wavelenghts 193 nm, 196 nm and 200 nm were tested, varying the bandwidth in the range 6 nm to 20 nm. With the selection criterion of possible high signal-to-noise ratio, the analytical wavelength 193 nm and bandwidth 6 nm were taken for further work. The injection volume was 6 μL and the column was termostated (20 °C).
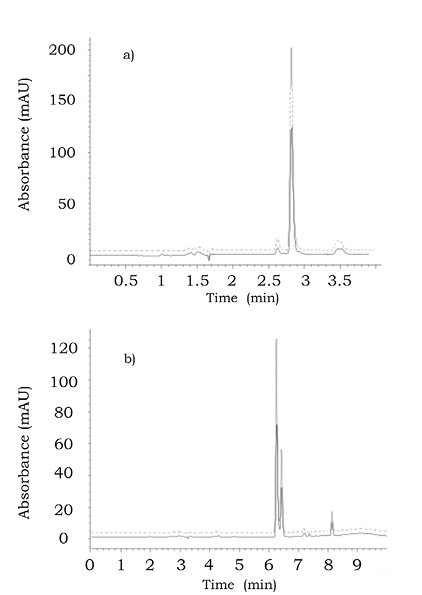
The analytical parameters evaluated for this procedure are summarized in table 1 and the chromatograms obtained in the analysis of real sample and this same sample after standard addition are presented in figure 1a. As can be observed, the retention time of glucosamine was 2.834 min (dead volume at 1.65 min) with acceptable reproducibility as demonstrated by low relative standard deviation (RSD) obtained for 10 non-succesive injections performed during one week (RSD < 0.1%). Owing to the simple chemical composition of glucosamine-based pharmaceutical formulations, the capacity factor 0.72 was considered acceptable. The peak purity measured between 2.78 min and 2.88 min was 999.1 ± 3.0 and 998.9 ± 3.3, for the standard solution and for the real sample respectively.
Calibration solutions were prepared to cover concentration range expected in ten times diluted solutions of pharmaceutical formulations; linear regression function was obtained with r2 = 0.9998, indicating good linearity (table 1). Within-day and between-day precisions presented in table 1 were evaluated as relative standard deviation for 10 succesive measurements and as RSD for 10 measurements performed during one week, respectively. The replicate measuremets were carried out using standard solutions 0.1 mg mL-1 and 0.9 mg mL-1, each of them prepared daily from this same stock glucosamine standard (9 mg mL-1). Slightly better precision was obtained for peak height measurements, so this signal mode could be used for routine applications. It should be noted that the analytical performance achieved in this simple chromatographic procedure, especially in terms of detection limit, precision and short time of analytical run, are comparable to those recently repoted for capillary electrophoresis with capacitively coupled contactless conductivity detector (DL = 30 μg mL-1, RSD < 1.9% and 3 min run time) (Campo, Campo, Ferlazzo, Vinci & Calatroni, 2001).
Tabla 1.
Analytical parameters evaluated for the proposed procedure (without derivatization) and for OPA method.
|
|
Parameter
|
Without derivatization
|
OPA method
|
|
Peak height
|
Peak area
|
Peak area
|
|
Retention time ± SD 1, min
|
2.834 ± 0.003
|
6.275 ± 0.013
|
|
Linear regression 2
|
A = 317.89 c + 1.76
|
A = 978.75 c -5.87
|
A = 4320.6 c – 4.94
|
|
Standard error of the slope
|
4.62
|
10.09
|
108.20
|
|
Standard error of the intercept
|
2.55
|
6.55
|
14.61
|
|
R2
|
0.9998
|
0.9998
|
0.9991
|
|
DL 3, µg mL-1
|
0.05
|
0.02
|
0.07
|
|
QL 4, µg mL-1
|
0.16
|
0.06
|
0.23
|
|
Within-day precision 5:
|
|
|
|
|
RSD (0.1 mg mL-1, n=10), %
|
0.42
|
0.60
|
0.86
|
|
RSD (0.9 mg mL-1, n=10), %
|
0.15
|
0.13
|
0.51
|
|
Between-day precision 6:
|
|
|
|
|
RSD (0.1 mg mL-1, n=10), %
|
0.96
|
1.02
|
0.86
|
|
RSD (0.9 mg ml-1, n=10), %
|
0.57
|
0.79
|
0.51
|
|
Recovery (0.1 mg mL-1) 7, %
|
100.8
|
100.4
|
100.8
|
|
Recovery (0.2 mg mL-1) 7, %
|
100.5
|
100.6
|
100.6
|
1 – evaluated for 10 non-succeeding injection (realized during one week);
2 – A = absorbance value, mAU, c = glucosamine concentration, mg mL-1;
3 – detection limit based on S/N 3:1;
4 – quantification limit based on S/N 10:1;
5 – 10 succeeding injection of this same solution;
6 – 10 not-succeeding injections of this same solution (realized during one week);
7 – evaluated for 3 replicates of standard addition 0.1 mg mL-1 and 0.2 mg mL-1 (see Materials and methods).
Source: Authors own elaboration.
Abrir
 |
1 – evaluated for 10 non-succeeding injection (realized during one week);
2 – A = absorbance value, mAU, c = glucosamine concentration, mg mL-1;
3 – detection limit based on S/N 3:1;
4 – quantification limit based on S/N 10:1;
5 – 10 succeeding injection of this same solution;
6 – 10 not-succeeding injections of this same solution (realized during one week);
7 – evaluated for 3 replicates of standard addition 0.1 mg mL-1 and 0.2 mg mL-1 (see Materials and methods).
Source: Authors own elaboration. Close |
To examine stability of sample solution, similar repetitive measurements were carried out during one week, while keeping the sample at 4 °C. For the peak height signal mode, the RSD value obtained for sample solution containing 0.1 mg mL-1 was 1.14% as compared to 0.96% evaluated for glucosamine standard solution at this same concentration. These results confirm good stability of both, sample and calibration solutions over at least one week period of time. For recovery checking, glucosamine standard was added to the sample solution, as described in Materials and methods. Doing so, the concentrations of standard added in the final solution were 0.1 mg mL-1 and 0.2 mg mL-1; as presented in tables 1 and 2, practically complete recovery was obtained in each case both, for peak height and peak area siglal modes.
Tabla 2.
Results of recovery experiments obtained for the proposed procedure, mean concentration values determined in sample solution are given with respective standard deviation values (n = 3).
|
|
Standard
added,
μg mL-1
|
Peak height signal mode
|
Peak area signal mode
|
|
Mean ± SD (μg mL-1)
|
Recovery (%)
|
Mean ± SD (μg mL-1)
|
Recovery (%)
|
|
0
|
313.1 ± 0.9
|
-
|
304.8 ± 2.6
|
-
|
|
100
|
413.9 ± 1.1
|
100.8
|
405.2 ± 1.9
|
100.4
|
|
200
|
514.1 ± 1.3
|
100.5
|
506.0 ± 2.1
|
100.6
|
Source: Authors own elaboration.
Abrir
|
Source: Authors own elaboration. Close |
It is generally accepted that the conformity between results obtained in the analysis of this same sample by means of different analytical procedures indicate that the “true” concentration value for the target species had been approached. O-phthalaldialdehyde (OPA) has been previously used for the derivatization of glucosamine (on –NH2) prior to reversed phase separation (Domínguez & Dunn, 1987; Eikenes et al., 2005; Nemati et al., 2007) and this procedure has been selected in the present work for comparativepurposes. The reaction was carried out in automatic mode, as described in Materials and methods. The separation and detection conditions are also listed in Materials and methods. Since the analysis of pharmaceutical formulations does not require high sensitivity, spectrophotometric detection (340 nm) was used. The calibration range was the same as for the procedure without derivatization, however all solutions were diluted with the mobile phase (1:50) prior to injection. In figure 1b, the chromatograms obtained for the solution of pharmaceutical formulation with and without standard addition are shown. It should be stressed that the chromatographic run time was 10 min, more than two times longer with respect to the procedure without derivatization (4 min). Furthermore, the elution of two anomers of the OPA-glucosamine product can be observed in figure 1b, in agreement with the previous reports (Eikenes et al., 2005). For quantification, the sum of the two peak areas was taken. The analytical parameters evaluated for OPA method are given in table 1. As can be observed, the recovery values were 100.8% and 100.6% for two levels of standard addition (0.1 mg mL-1 and 0.2 mg mL-1), confirming the accuracy of this procedure.
|
| |
 |
| |
Figure 1. Reversed phase chromatograms obtained for product 2 by the two procedures: a) without derivatization and b) with OPA derivatization. (—) the diluted sample and (----) after standard addition (8 μL 0.5 mg mL-1 glucosamine, final concentration in the injected solution 0.2 mg mL-1). For better comparison of the graphs, they have been offset on y axis.
Source: Authors own elaboration. |
 |
Figure 1. Reversed phase chromatograms obtained for product 2 by the two procedures: a) without derivatization and b) with OPA derivatization. (—) the diluted sample and (----) after standard addition (8 μL 0.5 mg mL-1 glucosamine, final concentration in the injected solution 0.2 mg mL-1). For better comparison of the graphs, they have been offset on y axis.
Source: Authors own elaboration. Close |
Three commercial products were analyzed using the two chromatographic procedures. En each case, five sub-samples were run and the results are shown in table 3. No statistically significant differences were found between results obtained by the procedure without derivatization and those obtained by OPA method (ANOVA), which confirms good accuracy of the determination. On the other hand, for the two pharmaceuticals, the results obtained were consistent with the values reported by the manufacturer; however, the glucosamine concentration found in product 1 corresponded to about 70 % of the value given on the label. This finding confirms previous study, in wich similar discrepancies were observed while analyzing 14 different commerial products available in Canada (Russell, Aghazadeh-Habashi & Jamali, 2002) and also indicates the need for routine analytical control of these products, in consistency with the recent comprehensive review (Aghazadeh-Habashi & Jamali, 2011).
Tabla 3.
Analytical results obtained for glucosamine in the commercial pharmaceuticals in the application of the proposed procedure (without derivatization) and OPA method.
|
|
Pharmaceutical formulation
|
Mean result ± SD, mg/tablet (n = 5)
|
Peak area signal mode
|
|
Without derivatization
|
OPA
|
|
Peak height
|
Peak area
|
Peak area
|
|
Product 1 (500 mg/tablet)
|
361.2 ± 0.8
|
346.1 ± 0.3
|
363.6 ± 3.7
|
|
Product 2 (300 mg/tablet)
|
313.1 ± 0.9
|
304.8 ± 2.6
|
306.5 ± 1.4
|
|
Product 3 (500 mg/tablet)
|
495.0 ± 1.2
|
477.0 ± 5.6
|
501.5 ± 1.0
|
Source: Authors own elaboration.
Abrir
|
Source: Authors own elaboration. Close |
CONCLUSION
In this work, the feasibility of reversed phase chromatographic determination of glucosamine in pharmaceutical products without pre-column derivatization was demonstrated in terms of sensitivity, within-day and between-day precision and also in the recovery experiments. For accuracy checking, three commercial products were analyzed by the procedure proposed and after derivatization of glucosamine with OPA; no statistically significant differences were detected between the results obtained in each procedure, confirming accuracy. The main features that make this procedure attractive for high throughput routine analyses, are the following: (1) no pre-column derivatization; (2) the use of typical reversed phase analytical column; (3) isocratic elution; (4) high peak purity with short retention time; (5) chromatpographic run completed in 4 min without necessity of column cleanning and equilibration; (6) good precision and accuracy with extremely simple procedure and low cost.
ACKNOWLEDGEMENTS
The financial support from Consejo Nacional de Ciencias y Tecnología (Conacyt), Mexico (project CB-2012-01-178553) is kindly acknowledged.
REFERENCES
Aghazadeh-Habashi, A. & Jamali, F. (2011). The glucosamine controversy; a pharmacokinetic issue. Journal of Pharmacy & Pharmaceutical Sciences, 14(2), 264-273.
Ander, B., Karlsson, A. & Ohrlund, A. (2001). Determination of heparin on intraocular lens surfaces by ion chromatography. Journal of Chromatography, 917(1-2), 105-110.
Anumula, K. R. (1994). Quantitative determination of monosaccharides in glycoproteins by high-performance liquid chromatogra1phy with highly sensitive fluorescence detection. Analytical Biochemistry, 220(2), 275-283.
Campo, G. M., Campo, S., Ferlazzo, A. M., Vinci, R. & Calatroni, A. (2001). Improved high-performance liquid chromatographic method to estimate aminosugars and its application to glycosaminoglycan determination in plasma and serum. Journal of Chromatography B Biomedical Sciences and Applications, 765(2), 151-160.
Chaisuwan, P., Kongprasertsak, T., Sangcakul, A., Smith, N. W., Nachapricha, D., Wilairat, P. & Uraisin, K. (2011). Direct injection of human serum and and pharmaceutical formulations for glucosamine determination by CE-C4D method. Journal of Chromatography B, 879(23), 2185-2188.
Cheng, X., & Kaplan, L. A. (2003). Simultaneous Analyses of Neutral Carbohydrates and Amino Sugars in Freshwaters with HPLC-PAD. Journal of Chromatography Science, 41(8), 434-438.
Chiusaroli, R., Piepoli, T., Zanelli, T., Ballanti, P., Lanza, M., Rovati, L. C. & Caselli, G. (2011). Experimental pharmacology of glucosamine sulfate. Hindawi Publishing Corporation International Journal of Rheumatology, doi:10.1155/2011/939265
Cohen, S. A. & De Antonis, K. M. (1994). Applications of amino acid derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate. Analysis of feed grains, intravenous solutions and glycoproteins. Journal of Chromatography A, 661(1-2), 25-34.
Distler, J. & Anguelouch, A. (2006). Evidence-based practice: review of clinical evidence on the efficacy of glucosamine and chondroitin in the treatment of osteoarthritis. Journal of the American Academiy of Nurse Practitioners, 18(10), 487-493.
Domínguez, L. M. & Dunn, R. S. (1987). Analysis of OPA-derivatized amino sugars in tobacco by high-performance liquid chromatography with fluorimetric detection. Journal of Chromatography Science, 25(10), 468-471.
Eikenes, M., Fongen, M., Roed, L. & Stenstrømc, Y. (2005). Determination of chitosan in wood and water samples by acidic hydrolysis and liquid chromatography with online fluorescence derivatization. Carbohydrate Polymers, 61(1), 29-38.
Gaonkar, P., Khanvilkar, V., Shettigar, R. & Gadgoli, C. (2006). Spectrophotometric method for determination of glucosamine in tablets. Indian Journal of Pharmaceutical Science, 68(1), 83-84.
Liang, Z., Leslie, J., Adebowale, A., Ashraf, M. & Eddington, N. D. (1999). Determination of the nutraceutical, glucosamine hydrochloride, in raw materials, dosage forms and plasma using pre-column derivatization with UV HPLC. Journal Pharmaceutical and Biomedical Analysis, 20(5), 807-814.
López-Cervantes, J., Sánchez-Machado, D. I. & Delgado-Rosas, K. E. (2007). Quantitation of glucosamine from shrimp waste using HPLC. Journal of Chromatography Science, 45(4), 195-199.
Nemati, M., Valizadeh, H., Ansarin, M. & Ghaderi, F. (2007). Development of a simple and sensitive high-performance liquid chromatography method for determination of glucosamine in pharmaceutical formulations. Journal of AOAC International, 90(2), 354-357.
Palace, G. P. & Phoebe Jr., C. H. (1997). Quantitative determination of amino acid levels in neutral and glucosamine-containing carbohydrate polymers. Analytical Biochemistry, 244(2), 393-403.
Qi, L., Zhang, S. F., Zuo, M. & Chen, Y. (2006). Capillary electrophoretic determination of glucosamine in osteoarthritis tablets via microwave-accelerated dansylation. Journal Pharmaceutical and Biomedical Analysis, 41(5), 1620-1624.
Ragle, R. L. & Sawitzke, A. D. (2012). Nutraceuticals in the management of osteoarthritis: a critical review. Drugs Aging, 29(9), 717-731. doi: 10.1007/s40266-012-0006-3
Russell, A. S., Aghazadeh-Habashi, A. & Jamali, F. (2002). Active ingredient consistency of commercially available glucosamine sulfate products. Journal of Rheumatology, 29(11), 2407-2409.
Shao, Y., Alluri, R., Mummert, M., Koetter, U. & Lech, S. (2004). A stability-indicating HPLC method for the determination of glucosamine in pharmaceutical formaulations. Journal of Pharmaceutical and Biomedical Analysis, 35(3), 625-631.
Soga, T. & Heiger, D. N. (1998). Simultaneous determination of monosaccharides in glycoproteins by capillary electrophoresis. Analytical Biochemistry, 261(1), 73-78.
Solomon, W. D. S., Kumar, R. A., Anand, P. R. V., Sivakumar, R. & Venkatnarayanan, R. (2010). Derivatized HPTLC method for simultaneous estimation of glucosamine and ibuprofen in tablets. Journal of Pharmaceutical Research and Health Care, 2(2), 156-162.
Vlad, S. C., LaValley, M. P., McAlindon, T. E. & Felson, D. T. (2007). Glucosamine for pain in osteoarthritis: why do trial results differ? Arthritis & Rheumatology, 56(7), 2267-2277.
Yu, Y., Cai, L., Zuo, M. & Duan, G. (2005). Precolumn derivatization liquid chromatography with mass spectrometry assay for the determination of glucosamine in small volume human plasma. Annali di Chimica, 95(9-10), 709-713.
Zhou, J. Z., Waszkuc, T. & Mohammed, F. (2004). Single laboratory validation of a method for determination of glucosamine in raw materials and dietary supplements containing glucosamine sulfate and/or glucosamine hydrochloride by high-performance liquid chromatography with FMOC-Su derivatization. Journal of AOAC International, 87(5), 1083-1092.
Zhou, J. Z., Waszkuc, T. & Mohammed, F. (2005). Determination of glucosamine in raw materials and dietary supplements containing glucosamine sulfate and/or glucosamine hydrochloride by high-performance liquid chromatography with FMOC-Su derivatization: collaborative study. Journal of AOAC International, 88(4), 1048-1058.
Zhu, X., Cai, J., Yang, J. & Su, Q. (2005). Determination of glucosamine in impure chitin samples by high-performance liquid chromatography. Carbohydrate Research, 340(10), 1732-1738.